
| Главная | ||||
| Advertisements | ||||
| Фармацевтические технологии и упаковка | ||||
| Медтехника. Лекарства, изделия медназначения. Дезсредства | ||||
| Стоматолог-практик | ||||
| Статьи Фармацевтические технологии и упаковка - Лекарства по GMP | ||||
| Статьи Медтехника. Лекарства, изделия медназначения. Дезсредства | ||||
| Подписка | ||||
| Рекламодателям | ||||
| Контакты | ||||
Обзор требований к гигиеническим зонам на фармацевтических и медицинских производствах
Томас Шрайнер, доктор фармацевтики, руководитель подразделения GMP-экпертизы Фармаплан Дойчланд ГмбХ, Германия С 1985 по 1988 год работал в концерне «Фрезениус», в т.ч. в должности руководителя производства. С 1989 является сотрудником Фармаплан и занимается консалтингом в области GMP. Доктор Шрайнер – один из постоянных соавторов проекта «Путеводитель GMP» издательства «Века» – Европейского аналога ISPE. Введение- Важной составляющей организации производства активных субстанций, лекарственных средств и медицинских продуктов в соответствии с GMP является концепция гигиенических зон. Эта концепция включает в себя следующие аспекты: - Определение гигиенических зон (в особенности, по параметрам микробиологии и содержания аэрозольных частиц в окружающей среде) - Потоки материалов и персонала, и концепция переходных шлюзов - Базовые требования к вентиляционной технике (напр., ступени фильтрации, концепция ступеней давления и т.д.) Вышеуказанные аспекты должны быть обязательно учтены при разработке планировки производства в соответствии с GMP. Далее мы приводим обзор требований европейских государственных инстанций, а также дополнительной рекомендательной документации, издаваемой этими инстанциями и другими организациями, которые необходимо учитывать при определении гигиенических зон для различных групп продуктов. Требования к гигиеническим зонам1. Активные субстанции Классические активные субстанции Требования к гигиеническим зонам можно найти в документе ICH (Международная конференция по гармонизации технических требований для регистрации лекарственных средств для человека) Q7A «Правила надлежащего производства активных субстанций», опубликованном в Европейском пространстве в виде Приложения 18 к Руководству по GMP ЕС. В этом документе в пункте 4.10 записано следующее:
В данной директиве в явном виде исключаются стерильные активные субстанции, начиная с этапа стерилизации. Для таких субстанций приводятся ссылки на требования национальных государственных инстанций в отношении гигиенических зон для стерильных лекарственных средств. Таким образом, можно сказать, что за исключением активных субстанций, задекларированных как стерильные, определение гигиенических зон (включая номенклатуру для каждой отдельной гигиенической зоны) по параметрам допустимого содержания аэрозольных частиц и микробиологической нагрузки, на основании вышеуказанных нормативных требований лежит в ответственности производителя активных субстанций. Активные субстанции, получаемые биотехнологическими методами В случае активных субстанций, получаемых биотехнологическими методами из прокариотов и эвкариотов, необходимо учитывать особые требования. Это, в особенности, касается вирусной безопасности конечного продукта, а также риска контаминации инородными организмами или инородными веществами (напр., инородными белками). Нормативной базой для данной группы продуктов является Приложение 2 Руководства по GMP ЕС «Производство биологических лекарственных средств для применения человеком». В Пункте 7 данного документа записано: «...Степень разделения, необходимая для предотвращения перекрестной контаминации, определяется характеристиками продукта и используемым оборудованием.»
При определении гигиенических требований и структуры помещений для производства и контроля качества активных субстанций, получаемых методом генной инженерии, необходимо учитывать требования Постановления о Безопасности Генных Технологий, в части применяемых степеней безопасности. На пространстве США для таких субстанций действует базовое руководство ISPE «Биофармацевтические средства» (ISPE Baseline Guide «Biopharmaceuticals»). В этом документе отражены взгляды администрации FDA США, но при этом документ не носит нормативный характер. 2. Лекарственные средства Требование адекватности концепции гигиенических зон записано в основном тексте Руководства по GMP ЕС. Так, в главе 3. «Помещения и оборудование» приведены следующие требования: 3.3 Освещение, температура, влажность и вентиляция должны быть надлежащими и обладать такими характеристиками, чтобы они ни напрямую, ни косвенно не оказывали негативного влияния… на лекарственное средство. 3.7 Помещения должны быть по возможности расположены таким образом, чтобы производство протекало по логически упорядоченным шагам в соответствии с порядком технологических операций и классами чистоты. В главе 5 приведено следующее требование: 5.19 Перекрестная контаминация должна предотвращаться с помощью подходящих технических или организационных мероприятий, например: a) производства в пространственно отделенных зонах .... b) подходящих шлюзов и вытяжных устройств... и т.д.
Для нестерильных лекарственных средств подобных указаний в Руководстве по GMP ЕС не имеется. По этой причине производители лекарственных средств должны самостоятельно определять гигиенические зоны, основываясь на потенциальном риске. Помощь в этом может оказать Памятка Центрального управления здравоохранения Федеративных земель по лекарственным средствам и медицинским продуктам «Инспектирование квалификации и валидации на фармацевтическом производстве и в контроле качества» (Aide Mйmoire der Zentralstelle der Lдnder fьr Gesundheitsschutz bei Arzneimitteln und Medizinprodukten „Inspektion von Qualifizierung und Validierung in pharmazeutischer Herstellung und Qualitдtskontrolle“). Этот документ устанавливает гигиенические зоны E и F для нестерильных лекарственных средств, однако определение этих зон основывается только на микробиологических показателях воздуха. Этот внутренний QM-документ немецких надзорных органов не является нормативным требованием для фармпроизводителей Германии, однако он отражает взгляды и ожидания немецких инспекторов. Для пространства США требования к гигиеническим зонам описаны в Руководстве для Промышленности «Стерильные лекарственные средства, произведенные асептическими методами» (Guidance for Industry «Sterile Drug Products Produced by Aseptic Processing») от Сентября 2004. Требования к гигиеническим зонам для производства нестерильных лекарственных средств на американском пространстве администрацией FDA США не установлены. Помимо этого, требования к гигиеническим зонам можно найти в Руководстве USP 28. Взгляды и ожидания инспекторов США, в том числе применительно и к нестерильным лекарственным средствам, отражены в различных Руководствах ISPE (Международного Общества Фармацевтического Инжиниринга), которые, однако, не обладают регулирующей силой. 3. Медицинские продукты Общие требования к гигиене рабочей среды в производстве медицинских продуктов установлены в Части 21 Кодекса Федеральных Норм (CFR), раздел 820.70, пункт G. Отдельные указания для европейского пространства можно найти в действующих Европейских Нормах (напр. EN ISO 13485:2000, пункт 4.9). Однако, эти указания очень общие, и производители медицинских продуктов придерживаются требований и общепринятой практики для лекарственных средств. Поэтому часто на производствах стерильных медицинских продуктов гигиенические зоны определены в соответствии с Приложением 1 Руководства по GMP ЕС. Заключение При проектировании новых или реконструкции уже существующих производств в первую очередь необходимо идентифицировать применяемую нормативную базу. После этого можно приступить к разработке эскиза планировки. На этой стадии очень полезно провести анализ разработанного варианта планировки в рамках документальной Экспертизы по GMP (GMP-Review) с привлечением ответственных технических специалистов, представляющих разные функциональные подразделения и дисциплины. Для этого часто привлекаются эксперты из других организаций. ООО «Фармаплан» 119034, Москва, Чистый пер., 6/1. тел.: +7 (095) 291 24 04 факс: +7 (095) 291 06 61 Pharmaplan Deutschland GmbH Borkenberg 14, P.O. Box 11 60 D-61440 Oberursel im Taunus, Germany . Tel.: +49 (6171) 970 4500 Fax: +49 (6171) 970 4501 |
 Начиная с этого выпуска компания Фармаплан открывает новую рубрику, где будут публиковаться статьи, освещающие различные аспекты GMP. Автором приведенной ниже статьи, опубликованной в нескольких европейских специализированных изданиях, в т.ч., в журнале «ChemManager», является один из ведущих экспертов GMP немецкого подразделения компании Фармаплан доктор фармацевтики Томас Шрайнер.
Начиная с этого выпуска компания Фармаплан открывает новую рубрику, где будут публиковаться статьи, освещающие различные аспекты GMP. Автором приведенной ниже статьи, опубликованной в нескольких европейских специализированных изданиях, в т.ч., в журнале «ChemManager», является один из ведущих экспертов GMP немецкого подразделения компании Фармаплан доктор фармацевтики Томас Шрайнер.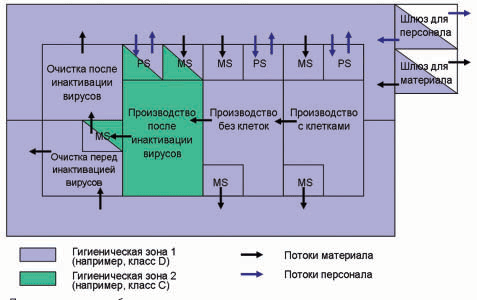 «Здания и оборудование, используемые для производства полупродуктов и активных субстанций, должны быть размещены, выполнены и сконструированы таким образом, чтобы они обеспечивали необходимый уровень очистки, обслуживания, а также соответствующую функциональность для данного типа и этапа технологического процесса...»
«Здания и оборудование, используемые для производства полупродуктов и активных субстанций, должны быть размещены, выполнены и сконструированы таким образом, чтобы они обеспечивали необходимый уровень очистки, обслуживания, а также соответствующую функциональность для данного типа и этапа технологического процесса...»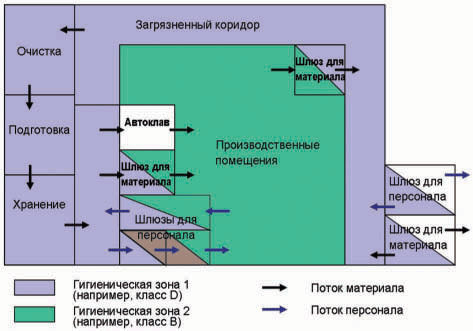 Помимо этого, в Германии необходимо учитывать требования проекта Памятки Центрального управления здравоохранения Федеративных земель по лекарственным средствам и медицинским продуктам «Биологическая и генная технология» (Aide Mйmoire der Zentralstelle der Lдnder fьr Gesundheitsschutz bei Arzneimitteln und Medizinprodukten «Bio- und Gentechnologie»). В этом проекте для различных технологических стадий даются рекомендации по гигиеническим зонам с соблюдением требований Приложения 1 Руководства по GMP ЕС. При этом необходимо учитывать, что этот документ был издан в 2004 году как проект, и дискутируется в фарминдустрии. Его можно рассматривать, как внутренний QM-документ европейских надзорных органов, определяющий их взгляд и ожидания от фармпроизводителей.
Помимо этого, в Германии необходимо учитывать требования проекта Памятки Центрального управления здравоохранения Федеративных земель по лекарственным средствам и медицинским продуктам «Биологическая и генная технология» (Aide Mйmoire der Zentralstelle der Lдnder fьr Gesundheitsschutz bei Arzneimitteln und Medizinprodukten «Bio- und Gentechnologie»). В этом проекте для различных технологических стадий даются рекомендации по гигиеническим зонам с соблюдением требований Приложения 1 Руководства по GMP ЕС. При этом необходимо учитывать, что этот документ был издан в 2004 году как проект, и дискутируется в фарминдустрии. Его можно рассматривать, как внутренний QM-документ европейских надзорных органов, определяющий их взгляд и ожидания от фармпроизводителей.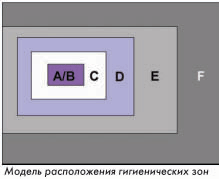 В Приложении 1 Руководства по GMP ЕС определены гигиенические зоны для конкретных технологических операций в производстве стерильных лекарственных средств. Они делятся по классам чистоты на зоны A, B, C и D, и отличаются друг от друга допустимым содержанием аэрозольных частиц и микробиологической нагрузкой для воздуха и поверхностей. Помимо этого в Приложении 1 можно найти детальные требования по ступеням фильтрации вентиляционного оборудования, переходным шлюзам, а также дифференциальному давлению между различными гигиеническими зонами.
В Приложении 1 Руководства по GMP ЕС определены гигиенические зоны для конкретных технологических операций в производстве стерильных лекарственных средств. Они делятся по классам чистоты на зоны A, B, C и D, и отличаются друг от друга допустимым содержанием аэрозольных частиц и микробиологической нагрузкой для воздуха и поверхностей. Помимо этого в Приложении 1 можно найти детальные требования по ступеням фильтрации вентиляционного оборудования, переходным шлюзам, а также дифференциальному давлению между различными гигиеническими зонами.| Дизайн webing.ru |